Computational Biology

The molecular history of Earth's biosphere
The DNA sequence of every organism on Earth contains historical information about its molecular past. Thus, genomic DNA sequences directly link natural history to biological chemistry. To develop those links, our group in 1990 became the first to comprehensively extract that history from a modern protein sequence database. With Gaston Gonnet, the group created the first web-accessible genomic analysis platform. This led to the MasterCatalog, a commercial database that used evolutionary history to organize genomic data. The MasterCatalog generated ca. $3.4 million in sales in its product lifetime.
This work also created bioinformatics tools to understand the roles in biology of biomolecules, connecting the genome to the organism, its diseases, its interaction with the environment, and its evolution. These set the stage for a planetary biology, where the history of Earth's biosphere is connected to the history of the planet and the cosmos.
The history of life on Earth is told in our DNA
Transforming our understanding of protein folding and function
Evolutionary analysis of protein sequence evolution provided the first convincing tools to predict the folded structure of proteins. The power of these tools was first illustrated by their prediction of the fold of protein kinase, before its crystal structure was solved. Further correct predictions were submitted to various structure prediction contests. Similar analyses allowed our group to publish the first examples of designed enzymes, where the mechanism for the enzyme was worked out. Based in part on this pioneering work, groups like those of David Baker are today designing proteins, and Deep Mind is using AI to predict protein folds.
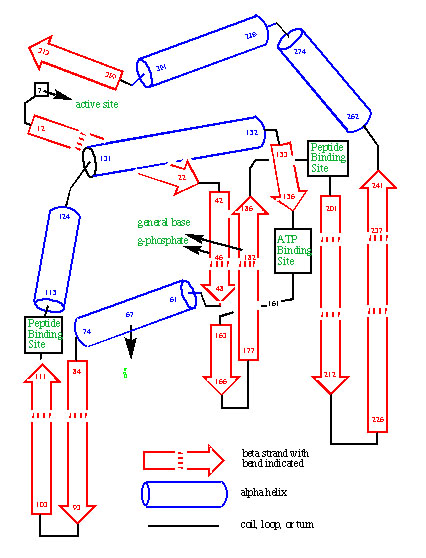
The prediction of the fold of protein kinase based on an analysis of the divergent evolution of the enzyme family. The prediction was published before an experimental structure was known.


