Evolutionary Bioinformatics Suite
The explosion of genomic sequence data over the past decade has created the need for tools to better connect the sequences of biological macromolecules to their fold, behavior and function. While convential annotation methods are sometimes helpful, they are insufficient to convey fully the information that is contained within genome sequence databases. It is also clear that the simplest evolution-based tools, often referred to as "comparative genomics", do not resolve many common annotation problems.
To overcome these deficiencies, the Foundation is developing a software suite (in collaboration with a molecular modeling company, Hypercube, Inc.) to examine genomic sequences using more sophisticated models within the context of their evolutionary history, with the expectation that more detailed insights can be gained about protein fold, behavior and function. This examination has come to be known as "phylogenomics". When defining phylogenomics, Jonathan Eisen observed that genomics, even in the version known as comparative genomics, had lagged behind other biological disciplines in exploiting the insights that are offered by the vast experiment that constitutes the three billion years of life on Earth.
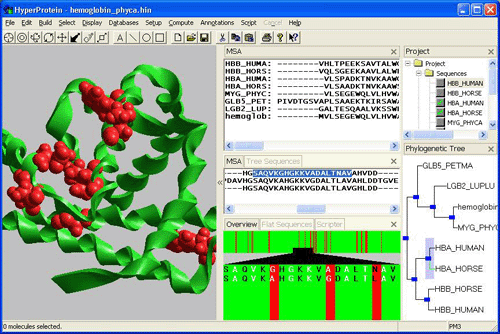
Phylogenomics seeks to rectify this through more complex analysis of evolutionary patterns. A phylogenomic analysis begins by identifying homologous genes, those related by common ancestry. It then seeks to place an evolutionary analysis of the family within a species context, identifying ortholog and paralog relationships. The analysis then examines patterns of amino acid replacement, the tempo of amino acid replacements, and the temporal sequence of events in gene evolution. The farther a phylogenomics analysis proceeds, the more information that can be extracted about fold, behavior, and function from the natural history of protein families.
Phylogenomic analyses are not limited to primary sequence data, of course. Similar principles can be extended to structures, pathways, and expression patterns. Phylogenomics has also been used to find key regulatory elements in non-coding genomic regions and delineate specificity determinants in proteins. More broadly, phylogenomic-types of analyses are offering fresh viewpoints in immunology, physiology, neurosciences, mental disease, and 'Darwinian medicine', which places human health and disease within an evolutionary perspective.
Phylogenomics can have practical implications for the pharmaceutical industry. For example, in the August 2003 issue of Nature Drug Discovery, David Searls, Head of the Bioinformatics Division of GlaxoSmithKline, discussed the concept of pharmacophylogenomics. Analysis of paralogs has been explored as a way to understand pathophysiology, seeking ways to move from biologically interesting but problematic targets to more tractable, druggable ones. The Foundation has been active in applied phylogenomics for several years. Indeed, Searls cites several papers that have emerged from the Foundation, and the laboratories of its consultants and collaborators, as important to pharmacophylogenomics. Some phylogenomic tools invented/developed at the Foundation, or in the laboratories of its consultants/collaborators, are currently being integrated into the HyperProtein bioinformatics suite in collaboration with Hypercube, Inc.


